
Given the ever-increasing prices of new drugs,1 the use of health technology assessment (HTA) by healthcare payers and insurers for guiding reimbursement and pricing decisions is becoming increasingly important for the allocation of limited resources. Clinical and economic data are analysed to judge the added clinical benefit and cost-effectiveness of new drugs versus existing treatments. At the same time, the market entry and patient access to new therapies have become politicised, with ongoing pressure to speed up drug evaluation processes, partially due to higher patient expectations and lobbying by the pharmaceutical industry.2 The evidentiary requirements for new drug approvals by regulatory agencies have been lowered, expanding the required evidence gap between regulatory approval and patient access, with the value ofmany new drugs surrounded by uncertainty at the point of reimbursement and pricing.3 New models of conditional access are emerging that involve additional data collection to reduce uncertainties in drugs’ clinical and cost-effectiveness.
In July 2021, NHS England announced the launch of the Innovative Medicines Fund (IMF) to fast-track promising new drugs, operating alongside and on similar terms to the Cancer Drugs Fund (CDF). In June2022, the IMF’s final principles specified a fixed annual budget of £340 m (equal to the CDF), creating a total funding pool of £680 m for early access to ‘the most promising’ medicines.4
From the CDF to the IMF
The CDF was launched in 2010 with a budget of £50m which spiralled to £340 m by 2015, without evidence of additional benefits to patients.5 This overspend triggered a review by the National Audit Office, which called into question the logic of the CDF, as no other condition had a ‘dedicated fund to provide access to drugs not routinely available on the National Health Service (NHS)’.6 As a result, in 2016, CDF was reformed to become a managed access fund for clinically uncertain drugs with the potential of satisfying criteria for routine use through the collection of new evidence within 2 years, while focusing on observational ‘real-world’ data (RWD).7 Following the reform, NHS England remained responsible for the administration of the fund but in close partnership with the National Institute for Health and Care Excellence(NICE), an ironic move given that the CDF was originally set up to fund drugs that NICE had already rejected. Early experience with the CDF suggests that its managed access model through further ‘real-world’ evidence or more mature data from the original randomised controlled trials (RCTs) has the potential to reduce clinical uncertainties.8–10 However, its value to society remains unproven with concerns about lack of transparency in drugs’ costs and time period during which they remain under the scheme.11 At the time ofthe 2016 CDF reform, commentators suggested that rather than relying on ‘real-world’ evidence to reduce uncertainty, funding for the CDF should be redeployed to undertake RCTs within routinely collected datasources.12 However, such suggestions for RCTs evaluating the relative cost-effectiveness of drugs in practice have not been followed, nor has the CDF made any major use of observational data. Instead, the reformed CDF has relied on examining more mature data from the original RCTs.13
Now, the CDF approach is expanded to non-cancer drugs to provide a similar opportunity for non-cancer patients to benefit from the latest potentially valuable treatments. Following a public consultation,14 eight guiding principles will shape the IMF, which are illustrated as part of the overall process listed in Figure 1.
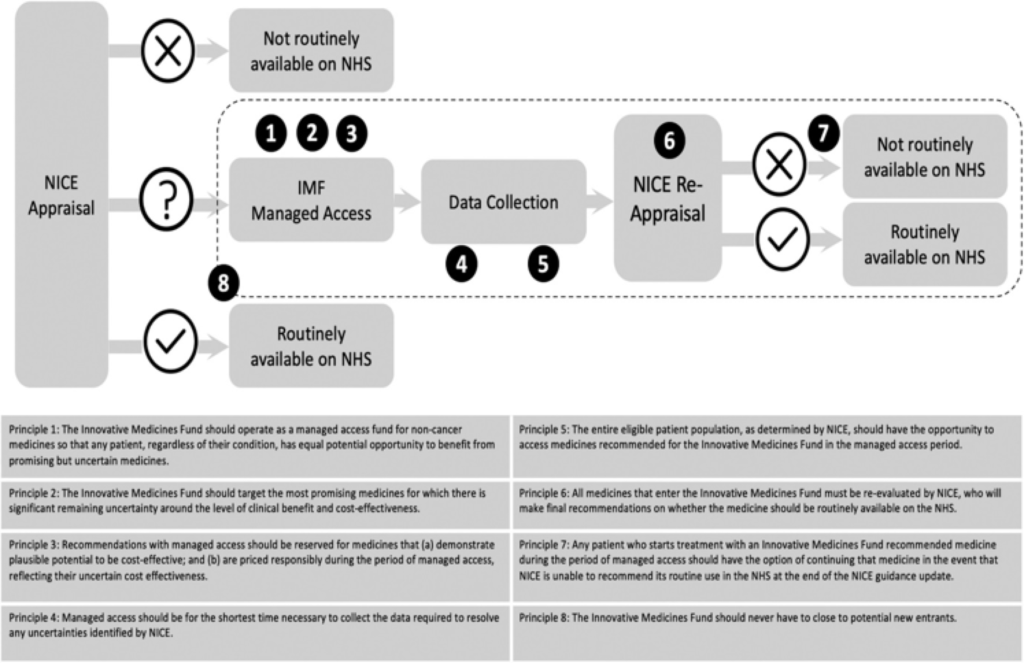
Figure 1. The newly launched IMF process and its guiding principles. Sources: Adapted based on NHS England Innovative Medicines Fund Principles,4 Kang and Cairns13 and NHS Commercial Framework for New Medicines.15 NICE: National Institute for Health and Care Excellence; NHS: National Health Service; IMF:Innovative Medicines Fund.
In general, we are broadly supportive of the eight guiding principles, although we believe their operationalisation is described in insufficient detail and without appropriate leveraging of the CDF experience. The IMF, similarly to the CDF, should be an exceptional route to patient access in case of immature evidence on drugs’ clinical and cost-effectiveness from RCTs, normally acting as the evidential gold standard. It should provide temporary reimbursement only while data collection activities are taking place to fill existing gaps, primarily via ongoing clinical trials in parallel with publicly funded observational research.
The ethical foundations: equal potential opportunity to benefit for all patients
We firmly agree that the ethical responsibility of the IMF should be to ensure that all patients, regardless oftheir condition, have an ‘equal potential opportunity to benefit from promising but uncertain medicines’, aswe do not believe it is justified, or fair, to provide such special funding arrangements only to cancer care.However, it is to be found out whether such an ‘equal potential opportunity’ will be the case for all non-cancer drugs, as the criteria for inclusion in the IMF might differ; although all new cancer drugs are ‘potentially eligible’ for CDF provision, it could be the case that only a subset of promising non-cancerdrugs will be considered for IMF. For example, the growing pipeline of treatments for ‘rare conditions’ is specifically highlighted, meaning that we are unclear as to whether an increased emphasis will be placed on orphan medicines in particular. Such a move might not be justified for the resource allocation of public funds, given the lack of evidence on such societal preferences for rare disease treatments in the United Kingdom16 and given the existing incentives afforded to rare-disease medicines elsewhere in their life-cycle.
At the same time, we do not see why such earmarked managed access schemes should only exist for medicines and no other types of interventions; for example, in the context of the CDF, no empirical evidence exists to support a ‘drugs-only’ rationale compared to other cancer care interventions such as surgery and radiotherapy, which seem to require the development of separate evidence-based value scales and reimbursement policies.17 The need to consider non-medicinal interventions might be particularly relevant in disease areas that lack effective pharmacological treatments, such as depression.18
Although the expansion of managed access arrangements to non-cancer medicines improves the ethicalsituation across disease areas, the ethical concerns now shift to types of interventions and whether thebalance between cancer and non-cancer treatment is appropriate. Nevertheless, we agree that the IMFshould operate alongside, and on similar (if not identical) terms to the CDF, allowing lessons to be drawnfrom the operation of the CDF to date. Indeed, we do not see any scientific rationale for having two separateschemes in place, so we suspect that after a suitable transition period, the schemes will be merged into asingle fund for all eligible medicines.
Entry criteria: ‘most promising’ needs better definition
We believe that the entry criteria for the ‘most promising’ medicines into the IMF are currently critically lacking in detail. To begin with, it seems that clinical uncertainty is exclusively related to efficacy/effectiveness only, with safety profile including toxicity not considered at all. Among the medicines’ entry criteria into IMF are to address ‘high unmet need’ and provide ‘significant clinical benefits’. In order to operationalise unmet need, it could be reasonable for drugs to meet the new disease severity modifier that has now replaced the EOL criteria; this is measured using the two metrics of total number of Quality Adjusted Life Years (QALYs) lost (‘absolute shortfall’) and fraction of QALYs lost(‘relative shortfall’) due to the condition. The magnitude of clinical benefit could become better defined using a scale, ideally QALYs as a generic health outcome measure to compare clinical benefits across therapeutic areas. If that is not practically possible, readily available scales based on disease-specific minimum clinical important differences19 or existing clinical value scales such as the European Society forMedical Oncology (ESMO) – Magnitude of Clinical Benefits Scale20 could be explored as second best alternatives.
Another entry requirement that needs better definition, relates to the representation of a ‘step-change in treatment for patients and clinicians’. At IMF entry, such a resolution will be challenging given the considerable uncertainty of medicines’ clinical benefits, and will likely only take place following the medicines’ utilisation in the community needed to reveal their effectiveness. A potential proxy for operationalising the step-change criterion at that time could be imposing a requirement for medicines to hold specific regulatory designations issued by the Medicines and Healthcare products Regulatory Agency(MHRA), such as the ‘Innovation Passport’ or the ‘Promising Innovative Medicine Designation’.
The final entry requirement for the new evidence generated to be considered ‘meaningful’ and ‘sufficiently reduce uncertainty’ should be viewed in the context of the fourth principle according to which the whole process ‘should be for the shortest time necessary to collect the data required to resolve any uncertainties’. More precisely, data should be collected within a ‘reasonable timeframe’, which will be defined on case-by-case basis but without exceeding 5 years (plus the NICE re-appraisal time), a sizeable extension to CDF’s time frame of 2 years. This is sensible as it represents more accurately the CDF timelines: out of 24 drugs that first exited the reformed CDF, the average time between the publication of the original and updated appraisals was 35.6 months (median of 36 months), with only 3 drugs being less than 2years.13 However, longer timeframes for such managed access schemes providing conditional reimbursement based on immature clinical evidence might act as a negative incentive for manufacturers to provide the necessary evidence required in the first place, e.g. a well-designed, sufficiently large, Phase 3 clinical trial, powered with the relevant meaningful outcomes. Longer timeframes also emphasise the need to specify the consequences of medicines remaining in the IMF beyond their recommended time period.NHS England could impose sanctions if data collection activities have not generated the required evidence and further time is needed, as for example taking the form of price discounts and rebates on sales.
Understanding managed access: the challenges of RWD
This last point takes us to the principle for the managed access schemes lasting the shortest time necessary to collect the required data for resolving the clinical uncertainties remaining. NICE will facilitate the development of the Data Collection Agreement (DCA) with reference to the appraisal committee’s considerations and will coordinate arrangements through a Managed Access Agreement (MAA) Oversight group with participation from NHS England, NHS Improvement, data custodians, the company, clinicians and patient groups, among others, and NICE will be responsible for maintaining a regular overview of eachDCA. On the other hand, the company will be responsible for producing a data/statistical analysis plan to ensure that methods and analytical outputs are outlined and agreed within 6 months of the MAA. However, based on the CDF experience, DCA multi-disciplinary groups seem to have only a cosmetic responsibility rather than real impact (typically requesting routinely collected data limited to a handful of time-to-event parameters); furthermore, details of the protocols, or statistical analysis plans, for the ‘real-world’ observational activities are rarely provided, nor what end points are used to evaluate the drugs, including the selection of patient-reported outcome measures.21 And in contrast to cancer audits and registries,22 which have a long history and are well understood, setting up new patient registries and collecting data for other diseases will be more challenging, with their timelines likely determined collaboratively between NHSEngland and the manufacturer.
We fully support the position that managed access should not replace the need for well-conducted RCTs and that ongoing trials should be the primary source of data, with RWD acting as a supplementary source to address evidence gaps. This would be aligned with the CDF experience, which although originally proposed as a ‘real-world’ initiative eventually became a vehicle for collecting more mature data from ongoing trials: out of 31 CDF drugs’ MAAs that had entered the CDF by July 2021, 26 (84%) specified that an ongoing trial would be the primary source of further data, with 24 of them requiring RWD as a secondary source; only 5 of the 31 MAAs (16%) specified RWD as a primary data source. An analysis of the first 24 drugs to exit the CDF by August 2022 highlights the limited role played by RWD and the importance of the original clinical trials’ longer follow-up.13
The main concern with observational studies is the absence of randomisation, which is needed to avoid bias in the estimates of relative effectiveness due to unadjusted differences in the characteristics of the individuals between the comparison groups, i.e. residual confounding. This and accompanying problems, such as immortal time bias, can be mitigated by trying to emulate a ‘target trial’, 23 or by adopting other statistical approaches such as propensity score matching.24 Such a framework for the emulation of observational data explicitly specifies key components of a hypothetical pragmatic ‘target trial’ (e.g.eligibility criteria, treatment assignment, start of follow-up),25 and can help with the assessment of risk of bias in non-randomised studies.26 Using the components of this framework, a recent study assessed the risk of bias in observational studies evaluating therapeutic interventions, with one-third of studies raising concerns because of unclear reporting or high risk of selection bias (confounding) and immortal time bias.27 This adds to other evidence showing the overall poor quality of RWD studies appraising the clinical benefits of approved therapies by the European Medicines Agency and US Food and Drug Administration,28 with only 2% of the 293 RWD studies appraised using population-based registry data. In this regard, the NHS hasno excuse for not using the administrative and clinical data at its disposal, including the various national clinical audit programmes.22
Non-superior and cost-ineffective drugs: safeguards needed against financial loss
Unsurprisingly, some of these promising new medicines will prove to be non-superior and/or cost-ineffective versus existing treatments, therefore reducing population health, given the health opportunity costs from their utilisation. If the IMF is to successfully foster early access to clinically effective, safe and cost-effective medicines, its operational details and mechanisms in place need to be carefully designed. Given the expanded time allowance of data collection activities, it becomes even more important to have the right safeguards so that decisions about ultimate rejections to routine commissioning do not come at the expense of other NHS patients. NHS England should more extensively consider the explicit use of confidential commercial arrangements,15 including the combination of budget caps with performance-based and outcomes-based agreements.29,30 This could help to protect the NHS against financial loss, in case that the new evidence demonstrates medicines to be clinically inferior or poor value-for-money.
This leads onto the seventh principle that ‘any patient who starts treatment with an IMF recommended medicine’ should have the ‘option of continuing that medicine’ in case NICE does not recommend it for routine use following re-evaluation. However, as acknowledged in the document, no additional funding will become available for medicines not recommended by NICE, so manufacturers will be financially responsible for patient access. It is not entirely clear whether this principle refers to treatment cost accrued during the IMF period or life-time costs following the exit from the IMF. However, the latter could provide better incentives: attract medicines with prospects on therapeutic superiority, possibly from innovative manufacturers willing to invest in high-risk projects for step-change improvements rather than minor or no therapeutic benefits.
There is an opportunity cost with every NHS decision
The final principle – that the IMF should not overspend its budget, nor should it close to new patients – is laudable but could prove challenging. For this, expenditure control mechanisms will have to be rigorously applied, given the excessive overspent experience with the original CDF.31 This highlights an overarching principle of HTA that is implicit in these arrangements and fundamental to the operation of NICE: that of opportunity cost. Setting up separate funds does not circumvent the healthcare system’s opportunity cost asthe establishment of both CDF and IMF is due to the mobilisation of additional resources that could, with the right political will, be used to fund existing, cost-effective care. Addressing unmet clinical needs is important, but the introduction of any new technology should be cost-effective so that it does not displace the benefits of existing effective and cost-effective care from other NHS patients. It is incumbent on theGovernment and NHS England to ensure that public funds dedicated to these initiatives are spent on faster and fairer access to clinical and cost-effective care (both drug and non-drug technologies), rather than to justify the high prices of drugs with weak or no evidence of therapeutic superiority and value for money. In doing so, the Government should use lay information campaigns to communicate to the wider patient and public community the underlying opportunity cost principle, by explaining that every additional pound spent on new technologies is likely to be found from displacing funding for existing health services.
Underlying comparative effectiveness issues and policy options
Evidence from regulatory approvals reveal that most new cancer drugs that entered the market at the beginning of the last decade had no evidence of benefit on hard clinical outcomes (i.e. survival or quality oflife).32 In the CDF, a minority of drugs were associated with significant overall survival benefit or satisfied the American Society of Clinical Oncology and ESMO clinical benefit scales criteria.5 In rare diseases, which could be relevant for the IMF, orphan drugs seem to be associated with larger median incremental health gains compared to non-orphan drugs, but with substantially higher costs and less favourable cost-effectiveness.33 Therefore, ensuring the value for money of such drugs would be critical for their routine commissioning to NHS.
The above raises concerns over whether implementing the IMF might incentivise the market entry of high-priced drugs for rare diseases with weak evidence on clinical benefits, such as personalised medicines for small patient populations. If the establishment of the IMF facilitates the funding of high priced drugs of uncertain value, such a managed access scheme risks disincentivising the generation of essential evidence, with managed access becoming the rule rather than the exception. This change would shift the timing of evidence generation from licensing to reimbursement, and the financial burden from the pharmaceutical industry to the public finances, at a time when opportunity costs would appear especially high.
For that purpose, protective safeguards should be in place throughout all IMF stages with public health objectives, relating to improvement of health outcomes, including at IMF entry and exit. Besides risk-sharing agreements to protect the NHS against financial loss in case a medicine proves to be clinically inferior or cost-ineffective following NICE re-evaluation, strict guidance should control the type of evidence used for IMF entry; for example, the consideration of non-randomised evidence should only be allowed based on appropriate justification, such as ethical reasons, while disregarding industry claims that small patient populations do not allow for such designs.
A more drastic solution would be to push back the responsibility of evidence generation for therapeutic superiority to the stage of regulatory approval.34 Imposing stricter evidence requirements on added clinical benefit versus existing comparators would incentivise manufacturers to conduct head-to-head randomised trials for establishing relative effectiveness.
Given that public taxpayer money is used to fund expensive treatments of uncertain value, data transparency becomes crucial, with important implications on credibility and fairness. Because of the intuitive uncertainty in their evidence base, once drugs enter the IMF and CDF, the full package of clinical and economic data submitted to MHRA and NICE forming the basis of MAA should become publicly available to allow for independent analysis by other groups, excluding any confidential commercial agreements relating to price discounts. Similarly, RWD generated as part of the IMF use, should also become publicly available and exposed to independent scrutiny.
In conclusion, the IMF, like the CDF, should be an exceptional route to patient access while providing the requisite evidence (mainly from RCTs) for reducing uncertainty about a drug’s clinical and cost-effectiveness. Potential inclusion in the IMF should be limited to those cases where the major uncertainties can be addressed within the defined timeframe. The notion of opportunity cost must not be ignored as IMF funding could always be used for other health services and technologies with strong evidence on effectiveness and value for money, which could improve overall population health.
Declarations
Competing Interests
Aris Angelis was a 2021–22 scholar at the National Institute for Health and Care Excellence and has had research grants from Novartis and Krystal Biotech, advisory fees from the Department of Health and Social Care and the European Commission, and shares ownership in a health tech company developing software tools for decision making. Andrew Briggs has acted as consultant to various commercial companies that have products funded through the NHS.
Funding
None declared.
Ethics approval
Not applicable. No ethical approval was required as no personal information was collected.
Guarantor
ArA and AB.
Contributorship
Conceptualisation and design: Angelis, Aggarwal, Miners, Grieve, Cairns, Briggs.Data collection: Angelis
Data analysis and interpretation: Angelis, Aggarwal, Miners, Grieve, Cairns, Briggs
Writing of the article:
Angelis, Miners, Briggs
Provenance
Not commissioned; peer reviewed by Laura Ward.
ORCID iD
Aris Angelis https://orcid.org/0000-0002-0261-4634
References
1. Rome BN, Egilman AC, Kesselheim AS. Trends in prescription drug launch prices, 2008–2021. JAMA 2022; 327:2145–2147.
2. Wouters OJ. Lobbying expenditures and campaign contributions by the pharmaceutical and health-product industry in the United States, 1999–2018. JAMA Intern Med 2020; 180: 688–697.
3. Cherla A, Naci H, Kesselheim AS, Gyawali B, Mossialos E. Assessment of coverage in England of cancer drugsq ualifying for US food and drug administration accelerated approval. JAMA Intern Med 2021; 181: 490–498.
4. NHSE. The Innovative Medicines Fund Principles. London: NHS England, 2022.
5. Aggarwal A, Fojo T, Chamberlain C, Davis C, Sullivan R. Do patient access schemes for high-cost cancer drugs deliver value to society? – lessons from the NHS Cancer Drugs Fund. Ann Oncol 2017; 28: 1738–1750.
6. National Audit Office. Investigation into the Cancer Drugs Fund. 2015.
7. NICE. PMG9 addendum – final amendments to the NICE technology appraisal methods guide to support the newCancer Drugs Fund arrangements, 2016. See www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-technology-appraisals/process-and-methods-guide-addendum.pdf (last checked 28 June 2023).
8. NICE. Obinutuzumab with Bendamustine for Treating Follicular Lymphoma after Rituximab. Technology appraisal guidance [TA629]. London: National Institute for Health and Care Excellence, 2020.
9. NICE. Nivolumab for Advanced Squamous Non-Small-Cell Lung Cancer after Chemotherapy. Technology appraisal guidance [TA655]. London: National Institute for Health and Care Excellence, 2020.
10. NICE. Nivolumab for Advanced Non-Squamous Non-Small-Cell Lung Cancer after Chemotherapy. Technology appraisal guidance [TA713]. London: National Institute for Health and Care Excellence, 2021.
11. Wood EM, Hughes DA. The new and non-transparent Cancer Drugs Fund. Pharmaco Economics 2020; 38: 1–4.
12. Grieve R, Abrams K, Claxton K, Goldacre B, James N, Nicholl J, et al. Cancer Drugs Fund requires further reform.BMJ 2016; 354: i5090.
13. Kang J, Cairns J. “ Don’t think twice, it’s all right”: using additional data to reduce uncertainty regarding oncologic drugs provided through managed access agreements in England. Pharmaco Economics – Open 2023; 7: 77–91.
14. NHS England. The Innovative Medicines Fund: engagement on proposals. 2021.
15. NHS England. NHS commercial framework for new medicines. 2021.
16. Linley WG, Hughes DA. Societal views on NICE, cancer drugs fund and value-based pricing criteria for prioritising medicines: a cross-sectional survey of 4118 adults in Great Britain. Health Econ 2013; 22:948–964.
17. Lievens Y, Audisio R, Banks I, Collette L, Grau C, Oliver K, et al. Towards an evidence-informed value scale for surgical and radiation oncology: a multi-stakeholder perspective. Lancet Oncol 2019; 20: e112–e23.
18. Cuijpers P, Stringaris A, Wolpert M. Treatment outcomes for depression: challenges and opportunities. Lancet Psychiatry 2020; 11: 925–927.
19. McGlothlin AE, Lewis RJ. Minimal clinically important difference: defining what really matters to patients. JAMA2014; 312: 1342–1343.
20. Cherny NI, Sullivan R, Dafni U, Kerst JM, Sobrero A, Zielinski C, et al. A standardised, generic, validated approach to stratify the magnitude of clinical benefit that can be anticipated from anti-cancer therapies: the European Society for Medical Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS). Ann Oncol 2015; 26: 1547–1573.
21. Macdonald H, Goldacre B. Does the reformed cancer drug fund generate evidence on effectiveness? A cross-sectional analysis on publicly accessible documentation. MedRxiv 2020.
22. HQPI. The National Clinical Audit Programme: healthcare quality improvement partnership, 2022. See https://www.hqip.org.uk/a-z-of-nca/#.Yofm-6jMI2w (last checked 28 June 2023).
23. Hernán MA, Sauer BC, Hernández-Díaz S, Platt R, Shrier I. Specifying a target trial prevents immortal time bias and other self-inflicted injuries in observational analyses. J Clin Epidemiol 2016; 79: 70–75.
24. Schöttker B, Saum K-U, Muhlack DC, Hoppe LK, Holleczek B, Brenner H. Polypharmacy and mortality: new insights from a large cohort of older adults by detection of effect modification by multi-morbidity and comprehensive correction of confounding by indication. Eur J Clin Pharmacol 2017; 73: 1041–1048.
25. Hernán MA, Robins JM. Using big data to emulate a target trial when a randomized trial is not available. Am J Epidemiol 2016; 183: 758–764.
26. Sterne JAC, Hernán MA, Reeves BC, Savović J, Berkman ND, Viswanathan M, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016; 355: i4919
27. Nguyen VT, Engleton M, Davison M, Ravaud P, Porcher R, Boutron I. Risk of bias in observational studies using routinely collected data of comparative effectiveness research: a meta-research study. BMC Med 2021; 19: 279.
28. Boyle JM, Hegarty G, Frampton C, Harvey-Jones E, Dodkins J, Beyer K, et al. Real-world outcomes associated with new cancer medicines approved by the Food and Drug Administration and European Medicines Agency: a retrospective cohort study. Eur J Cancer 2021; 155: 136–144.
29. Kanavos P, Ferrario A, Tafuri G, Siviero P. Managing risk and uncertainty in health technology introduction: the role of managed entry agreements. Global Policy 2017; 8: 84–92.
30. Carlson JJ, Chen S, Garrison LP. Performance-based risk-sharing arrangements: an updated international review. PharmacoEconomics 2017; 35: 1063–1072.
31. Angelis A, Lomas J, Woods B, and Naci H. Promoting population health through pharmaceutical policy: The role of the UK Voluntary Scheme, London School of Economics and Political Science, 2023. https://www.lse.ac.uk/lse-health/assets/documents/Reports/23-0275-Pharma-Report-V10.pdf
32. Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009–13. BMJ 2017; 359: j4530.
33. Chambers JD, Silver MC, Berklein FC, Cohen JT, Neumann PJ. Orphan drugs offer larger health gains but less favorable cost-effectiveness than non-orphan drugs. J Gen Int Med 2020; 35: 2629–2636.
34. Angelis A, Polyakov R, Wouters O, Torreele E, McKee M. High drug prices are not justified by industry’s spending on research and development. BMJ 2023; 380: e071710.
Aris Angelis1,2, Ajay Aggarwal1, Alec Miners1, Richard Grieve1, John Cairns1, and AndrewBriggs1
1 Department of Health Services Research and Policy, London School of Hygiene & Tropical Medicine, London, WC1H 9SH, UK
2 Department of Health Policy and LSE Health, London School of Economics and Political Science, London, WC2A 2AE, UK
Corresponding author(s): Aris Angelis. Email: [email protected]